 18928299212(微信同号) 0769-27280456
18928299212(微信同号) 0769-27280456
18928299212(微信同号) 0769-27280456

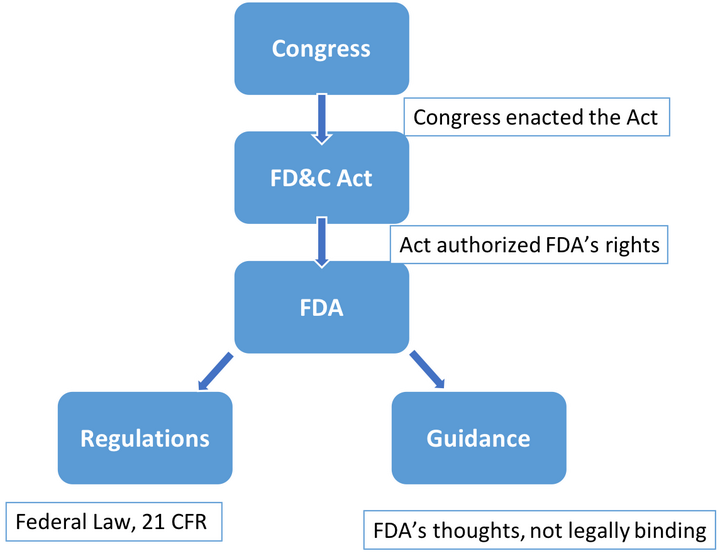
FDA, Food and Drug Administration,美国药监局,是法律赋予下负责监管美国药物、生物制剂、医疗器械、美容产品和烟草等的联邦机构。在讲FDA的权利来源时,不得不提到的就是FD&C Act(法案),由国会制定,赋予了FDA了监管一系列产品的权力。
2. 医疗器械的分类
在確定风险分类前,第一步肯定是要确定你的产品到底是不是器械(这不是废话~┑( ̄Д  ̄)┍,不是的)。医疗器械的定义,在不同的地域或者国家有不同的定于,特别是那种用途介于美容康复产品和器械之间的,就有可能出现这种情况。
风险分类:按照在临床使用中,器械可能对病人或者使用者带来的的风险等级的不同,和在这个基础上药监局制定的不同监管要求,来确定器械最终属于哪一个级别。I类为最低风险,III类为最高风险。
器械风险类别
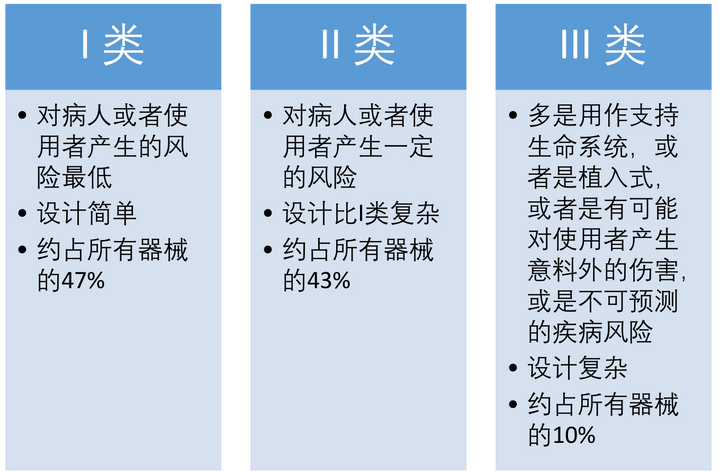
如果你一开始并不清楚到底你的器械所的风险类别,可以通过参考类似的产品来确定,方法有以下几种:
a. 不知道product code的情況下,可以通过输入器械的关键字,例如“pacemaker”去查收,https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm, 网站会列出所有在这关键字下的产品编码(product code),再根据描述选择对应的product code。在点击进去看详细内容前,就可以在右边看到这类型的产品属于哪一个产品类别。
b. 第二个就是直接查找器械对应的法规,这个就需要你比较了解器械或者是现有的器械法规分类的情况下用比较好,如果是不清楚的话,还是建议通过关键字查找。
3. 医疗器械的审批方式
器械申报途径
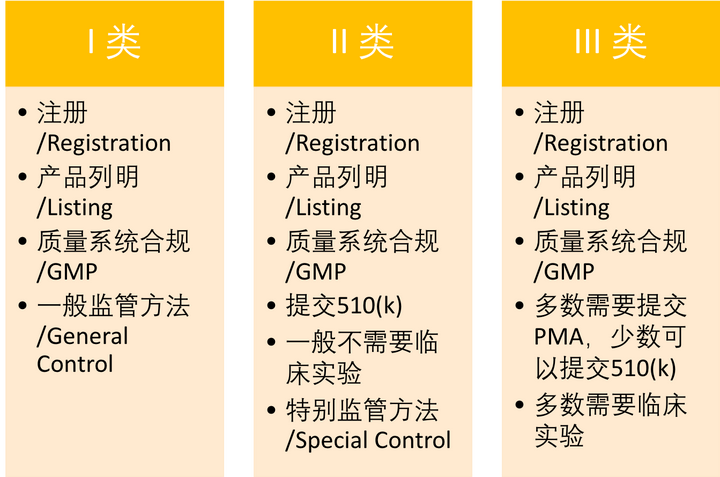
申报流程的选择,根据器械的类别和监管方式来决定。
FD&C 法案中的第510(k) 条款规定,器械生产商在美国销售产品前必须向FDA注册、提交审批文件,如果不需要提交PMA,又不符合510(k)豁免资格的厂商,需要提交510(k),也即是Premarket Notification,PMN,以获得FDA的市场准入批准授函(Clearance)。对于提交上来的510(k)申请,厂商的目的是要向FDA证明他们的器械和现存的用作比较的器械(Predicate Device),至少具有一样的安全性和有效性,也就是“substantially equivalent”。至于怎样算是SE,FDA有给出了具体的要求:
由此我们看出“same intended use” 和”technological characteristics”是FDA关注的两个重点,是FDA做出SE与否的关键因素;对于厂家来说,选择合适的参照器械,和好好准备文件针对这些关键,也是他们能不能比较顺利并且在较短的时间内拿到审批决定的重点。
而对于第III类别的器械,FDA认为一般的和特别的监管方法都不能很好的保证器械的安全和效
用,因此这类别的器械就需要提交上市前允许的审批申请,也就是Premarket Approval,简称
PMA,它也是FDA规定的最严格的上市审批方式。相对于510(k)只需要证明和参照器械有“实质等同”,PMA要求商家要提供足够的科学证据来证明他们生产的器械是安全,和有效的,这就变成了多数时候,厂家必须提供相当充分的临床实验和数据,来满足审批要求。
要注意的是,需要提交PMA的器械有些是涉及到新的内容或者是设计,FDA有可能还没有建立起针对这系列器械的法规要求,那官网上能找到的只有器械种类和产品编码的信息,没有风险类别。当商家的器械满足高风险的内容,而又没有找到资料清晰记录需要提交哪一种审批的话,厂家可以通过和FDA进行提交前会议,513(g),搜索PMA和510(k)数据库等的方式,找到相关信息。




